
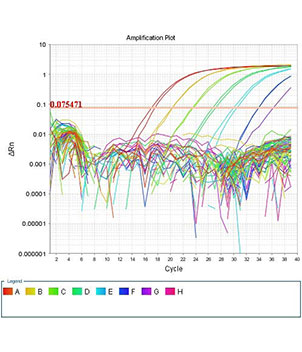

برخی از تکنیک های ارائه شده در هیستوژنوتک
پیشرفت های دو دهه اخیر در زمینه بیولوژی مولکولی موجب ارتقاء کارآیی و تکامل هر چه بیشتر آزمایش های وابسته به DNA در علوم پزشکی گردیده است.
بحث عمده در زیستشناسی مولکولی، استنباط برهم کنش بین سیستمهای درون سلولی ( از جمله برهمکنشهای RNA ،DNA و پروتئین سازی) و چگونگی تنظیم این برهمکنشها می باشد که مورد بررسی قرار میگیرد.
برخی از تکنیک های ارائه شده در هیستوژنوتک
پیشرفت های دو دهه اخیر در زمینه بیولوژی مولکولی موجب ارتقاء کارآیی و تکامل هر چه بیشتر آزمایش های وابسته به DNA در علوم پزشکی گردیده است.
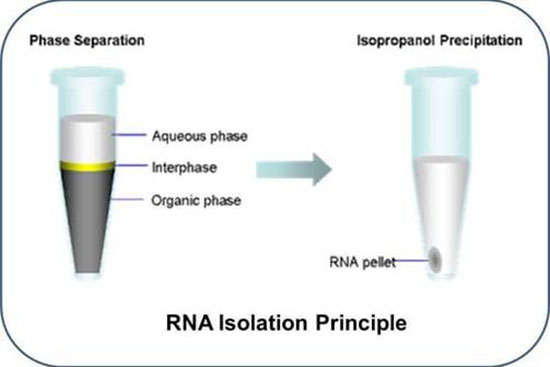
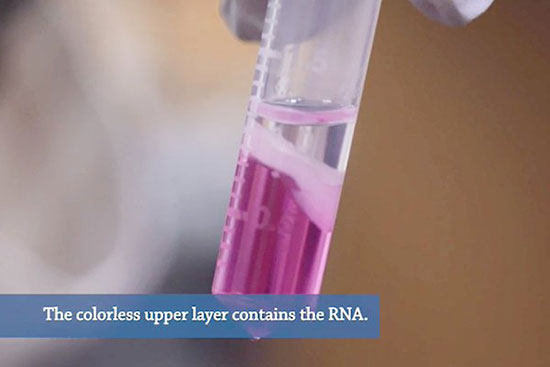
استخراج آر ان ای از نمونه های بیولوژیکی اولین گام اساسی در مطالعه بسیاری از فرآیندهای بیولوژیکی است. RNA نقش کلیدی در بیان و تنظیم ژن ایفا می کند. فرآیند استخراج RNA به منظور بیان ژن با همگن سازی نمونه و سپس لیز سلول ها برای آزادسازی RNA شروع شده و به دنبال آن شستشو و تخلیص از نمک ها و سایر آلاینده ها با استفاده از سانترفیوژ دور بالا صورت می گیرد. در هیستوژنوتک استخراج RNA کل و انواع اختصاصی RNA (microRNA) به دو روش دستی (فنل کلروفورم(ترایزول) ) و کیت از نمونه های مختلف زیستی (سلول ، تخمک، بافتهای سخت و نرم، بافت های گیاهی و…) با حداکثر غلظت و کیفیت قابل انجام می باشد. به منظور تایید کیفیت RNA استخراج شده با استفاده از نانودراپ درصد خلوص و غلظت آن کنترل شده و آماده استفاده برای بررسی بیان ژن، توالی یابی کل ژنوم (NGS) و … خواهد شد.
استخراج آر ان ای از نمونه های بیولوژیکی اولین گام اساسی در مطالعه بسیاری از فرآیندهای بیولوژیکی است. RNA نقش کلیدی در بیان و تنظیم ژن ایفا می کند. فرآیند استخراج RNA به منظور بیان ژن با همگن سازی نمونه و سپس لیز سلول ها برای آزادسازی RNA شروع شده و به دنبال آن شستشو و تخلیص از نمک ها و سایر آلاینده ها با استفاده از سانترفیوژ دور بالا صورت می گیرد. در هیستوژنوتک استخراج RNA کل و انواع اختصاصی RNA (microRNA) به دو روش دستی (فنل کلروفورم(ترایزول) ) و کیت از نمونه های مختلف زیستی (سلول ، تخمک، بافتهای سخت و نرم، بافت های گیاهی و…) با حداکثر غلظت و کیفیت قابل انجام می باشد. به منظور تایید کیفیت RNA استخراج شده با استفاده از نانودراپ درصد خلوص و غلظت آن کنترل شده و آماده استفاده برای بررسی بیان ژن، توالی یابی کل ژنوم (NGS) و … خواهد شد.
بررسی میزان بیان ژن ها درنمونه های زیستی، نیازمند تعیین بررسی تعداد کپی های آن ژن در سطح mRNA می باشد. از طرفی اساس فرآیند PCR برای بیان ژن، تکثیر DNA است. از این رو لازم است ابتدا RNA با استفاده از آنزیم نسخه برداری معکوس به DNA تبدیل شود که از آن تحت عنوان cDNA (Complementry DNA) نام برده می شود. واکنش نسخه برداری معکوس (سنتز cDNA) را می توان بر روی RNA کل سیتوپلاسمی و یا بر روی mRNA انجام داد. در هیستوژنوتک سنتز cDNA با استفاده از کیت های تک مرحله ای یا چند مرحله ای همراه با پرایمر های عمومی (Random hexamer- Oligo dT) و یا پرایمر های اختصاصی هر ژن ( microRNA stem loop) در سیکل دمایی منظم، در دستگاه ترموسایکر انجام می شود.
بررسی میزان بیان ژن ها درنمونه های زیستی، نیازمند تعیین بررسی تعداد کپی های آن ژن در سطح mRNA می باشد. از طرفی اساس فرآیند PCR برای بیان ژن، تکثیر DNA است. از این رو لازم است ابتدا RNA با استفاده از آنزیم نسخه برداری معکوس به DNA تبدیل شود که از آن تحت عنوان cDNA (Complementry DNA) نام برده می شود. واکنش نسخه برداری معکوس (سنتز cDNA) را می توان بر روی RNA کل سیتوپلاسمی و یا بر روی mRNA انجام داد. در هیستوژنوتک سنتز cDNA با استفاده از کیت های تک مرحله ای یا چند مرحله ای همراه با پرایمر های عمومی (Random hexamer- Oligo dT) و یا پرایمر های اختصاصی هر ژن ( microRNA stem loop) در سیکل دمایی منظم، در دستگاه ترموسایکر انجام می شود.
استخراج دی ان ای اغلب یک گام اولیه در بسیاری از فرآیندهای تشخیصی است که برای تشخیص انواع باکتری ها، ویروس ها در محیط، همچنین تشخیص بیماری ها و اختلالات ژنتیکی استفاده می شود. استخراج DNA شامل لیز سلول ها و حل شدن DNA می باشدکه با روش های شیمیایی یا آنزیمی برای حذف ماکرومولکول ها، لیپیدها، RNA و یا پروتئین ها همراه خواهد بود. تکنیک های رایج برای استخراج DNA شامل استخراج آلی (روش فنل -کلروفرم)، روش غیرآلی (نمک زدایی و تیمار با پروتئیناز K) و روش جذب (غشای ژل سیلیکا) می باشد. در هیستوژنوتک با استفاده از روش های دستی و همچنین کیت های تجاری موجود، استخراج DNA از بافت های مختلف از جمله خون، مایعات بدن، بافت های تعبیه شده در پارافین یا فرمالین، بافت های منجمد و نمونه های باستانی و… صورت می گیرد.
استخراج دی ان ای اغلب یک گام اولیه در بسیاری از فرآیندهای تشخیصی است که برای تشخیص انواع باکتری ها، ویروس ها در محیط، همچنین تشخیص بیماری ها و اختلالات ژنتیکی استفاده می شود. استخراج DNA شامل لیز سلول ها و حل شدن DNA می باشدکه با روش های شیمیایی یا آنزیمی برای حذف ماکرومولکول ها، لیپیدها، RNA و یا پروتئین ها همراه خواهد بود. تکنیک های رایج برای استخراج DNA شامل استخراج آلی (روش فنل -کلروفرم)، روش غیرآلی (نمک زدایی و تیمار با پروتئیناز K) و روش جذب (غشای ژل سیلیکا) می باشد. در هیستوژنوتک با استفاده از روش های دستی و همچنین کیت های تجاری موجود، استخراج DNA از بافت های مختلف از جمله خون، مایعات بدن، بافت های تعبیه شده در پارافین یا فرمالین، بافت های منجمد و نمونه های باستانی و… صورت می گیرد.
افزایش سریع مقاومت آنتی بیوتیکی، منجر به مطالعات بیشتر در مورد عناصر ژنتیکی متحرک (مانند پلاسمید) شده است. پلاسمیدها ناقل اصلی اشتراک ژن، در بین باکتری ها هستند. بنابراین نقش کلیدی در تکامل میکروبی و گسترش مقاومت آنتی بیوتیکی ایفا می کنند. به همین دلیل مطالعه در سطح پلاسمید به دلیل افزایش مقاومت آنتی بیوتیکی در باکتری های بیماری زا، بین گونه های مختلف حیوانی و انسانی بسیار قابل اهمیت می باشد. چون پلاسمید بخش کوچکی از DNA کل را شامل می شود، استخراج آن نیاز به فرآیند تکثیر باکتری در انکوباتور و سپس تخلیص دارد. در هیستوژنوتک فرآیند های تکثیر، استخراج و تخلیص پلاسمید با استفاده از کیت های اختصاصی و روش های دستی، قابل انجام می باشد.
افزایش سریع مقاومت آنتی بیوتیکی، منجر به مطالعات بیشتر در مورد عناصر ژنتیکی متحرک (مانند پلاسمید) شده است. پلاسمیدها ناقل اصلی اشتراک ژن، در بین باکتری ها هستند. بنابراین نقش کلیدی در تکامل میکروبی و گسترش مقاومت آنتی بیوتیکی ایفا می کنند. به همین دلیل مطالعه در سطح پلاسمید به دلیل افزایش مقاومت آنتی بیوتیکی در باکتری های بیماری زا، بین گونه های مختلف حیوانی و انسانی بسیار قابل اهمیت می باشد. چون پلاسمید بخش کوچکی از DNA کل را شامل می شود، استخراج آن نیاز به فرآیند تکثیر باکتری در انکوباتور و سپس تخلیص دارد. در هیستوژنوتک فرآیند های تکثیر، استخراج و تخلیص پلاسمید با استفاده از کیت های اختصاصی و روش های دستی، قابل انجام می باشد.
مولکول های پرایمر، اولیگودئوکسی نوکلئوتید یا به عبارتی DNA تک رشته می باشند که به طور اختصاصی برای منطقه مشخصی از DNA هدف ساخته می شوند. پس از هیبریدیزاسیون پرایمر در ناحیه مکمل، سنتز DNA از انتهای OH پرایمر ادامه می یابد. طراحی آگاهانه پرایمر در قلب هر پروژه تحقیقاتی می باشد. یک پرایمر ضعیف منجر به ایجاد محصول غیر اختصاصی و در نهایت نتایج اشتباه خواهد شد. نرم افزارهای متنوع تحت وب برای به حداقل رساندن اشتباهات در روند طراحی پرایمر ایجاد شده است که در هیستوژنوتک با بهره گیری از دانش فنی نیروهای مجرب، انواع پرایمر ها exon-exon junction) و (…, intron-spanning جهت بررسی بیان ژن به روش سایبرگرین و کلاسیک وجود دارد. پس از طراحی پرایمر، کیفیت پرایمر ها با استفاده از نرم افزارهای رایج (Primer3,UCSC,Gene Runner, Oligo7,Primer Blast) کنترل، سپس سنتز پرایمر انجام خواهد شد. کارایی پرایمر های ساخته شده در محیط آزمایشگاه با ایجاد سریال رقت و رسم منحنی استاندارد و همچنین کنترل سایز محصولات حاصل از آن با استفاده از ژل الکتروفورز صورت می گیرد.
مولکول های پرایمر، اولیگودئوکسی نوکلئوتید یا به عبارتی DNA تک رشته می باشند که به طور اختصاصی برای منطقه مشخصی از DNA هدف ساخته می شوند. پس از هیبریدیزاسیون پرایمر در ناحیه مکمل، سنتز DNA از انتهای OH پرایمر ادامه می یابد. طراحی آگاهانه پرایمر در قلب هر پروژه تحقیقاتی می باشد. یک پرایمر ضعیف منجر به ایجاد محصول غیر اختصاصی و در نهایت نتایج اشتباه خواهد شد. نرم افزارهای متنوع تحت وب برای به حداقل رساندن اشتباهات در روند طراحی پرایمر ایجاد شده است که در هیستوژنوتک با بهره گیری از دانش فنی نیروهای مجرب، انواع پرایمر ها exon-exon junction) و (…, intron-spanning جهت بررسی بیان ژن به روش سایبرگرین و کلاسیک وجود دارد. پس از طراحی پرایمر، کیفیت پرایمر ها با استفاده از نرم افزارهای رایج (Primer3,UCSC,Gene Runner, Oligo7,Primer Blast) کنترل، سپس سنتز پرایمر انجام خواهد شد. کارایی پرایمر های ساخته شده در محیط آزمایشگاه با ایجاد سریال رقت و رسم منحنی استاندارد و همچنین کنترل سایز محصولات حاصل از آن با استفاده از ژل الکتروفورز صورت می گیرد.
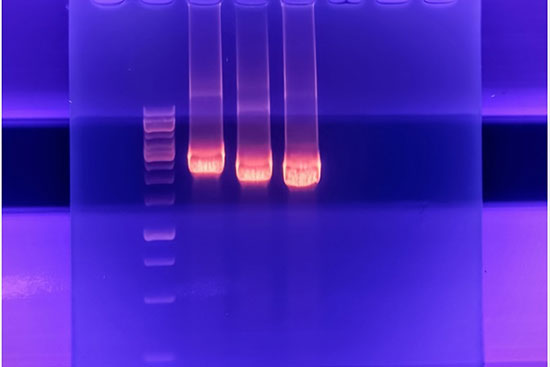
بررسی بیان ژن به روش پی سی آر کلاسیک، یکی از پرکاربردترین روش ها برای تجزیه و تحلیل بیان ژن است که در آن با استفاده از الکتروفورز ژل آگارز، محصول PCR مورد ارزیابی قرار می گیرد. مزیت این روش نسبت به qPCR بررسی محصولات DNA بر اساس اندازه و بار می باشد. الکتروفورز ژل آگاروز ساده ترین روش برای تجزیه و تحلیل محصولات PCR است. در این روش اندازه گیری سایز محصول PCR با استفاده از لدر انجام می گیرد. در هیستوژنوتک از این روش برای بررسی بیان ژن، تشخیص انواع سویه های باکتری، ویروس، گونه های جهش یافته و … صورت می گیرد. محصولات حاصل از این روش پس از انجام الکتروفورز در تابش نور UV در دستگاه ترنسلومیناتور و یا ژل داک قرار گرفته و باند ها از نظر تراکم، سایز و … با استفاده از نرم افزاز Image j مورد ارزیابی و آنالیز قرار می گیرند.
بررسی بیان ژن به روش پی سی آر کلاسیک، یکی از پرکاربردترین روش ها برای تجزیه و تحلیل بیان ژن است که در آن با استفاده از الکتروفورز ژل آگارز، محصول PCR مورد ارزیابی قرار می گیرد. مزیت این روش نسبت به qPCR بررسی محصولات DNA بر اساس اندازه و بار می باشد. الکتروفورز ژل آگاروز ساده ترین روش برای تجزیه و تحلیل محصولات PCR است. در این روش اندازه گیری سایز محصول PCR با استفاده از لدر انجام می گیرد. در هیستوژنوتک از این روش برای بررسی بیان ژن، تشخیص انواع سویه های باکتری، ویروس، گونه های جهش یافته و … صورت می گیرد. محصولات حاصل از این روش پس از انجام الکتروفورز در تابش نور UV در دستگاه ترنسلومیناتور و یا ژل داک قرار گرفته و باند ها از نظر تراکم، سایز و … با استفاده از نرم افزاز Image j مورد ارزیابی و آنالیز قرار می گیرند.
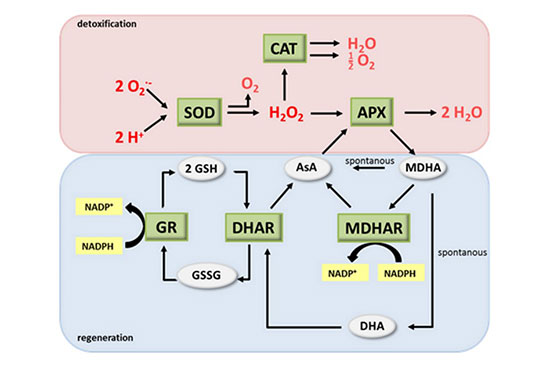
استرس اکسیداتیو عدم تعادل بین مولکولهای اکسیدان و آنتی اکسیدان، به نفع اکسیدانها است که باعث پیری و بیماری میشود. آنتی اکسیدانها یا عوامل ضد اکسیداسیون، با اتصال به عوامل استرس زا، اثر اکسیدانهای خطرناک را کاهش داده و قدرت تخریب آنها را کم میکنند. به همین دلیل سنجش میزان آنتی اکسیدانها، از ارزش بالایی برخوردار است که این ترکیبات در هیستوژنوتک با استفاده از کیتهای اختصاصی مورد ارزیابی قرار میگیرد. به طور مثال سنجش سوپراکسید دیسموتاز (SOD)، کاتالاز (CAT)، گلوتاتیون پراکسیداز (GPX) و همچنین برخی از آنتی اکسیدانها (مانند GSH) به روش غیر آنزیمی و با استفاده از ترکیبات شیمیایی در این مرکز مورد بررسی قرار می گیرد.. آسیبهای ناشی از عوامل استرس زا در سه سطح قابل بررسی است: ۱-آسیب پروتئینی، ۲-آسیب در متابولیسم چربی ۳-آسیب به DNA . محتوای پروتئین کربونیل (PC) یک نشانگر رایج جهت بررسی آسیب در سطح پروتئین است. پراکسیداسیون لیپیدها (اسیدهای چرب)معمولاً به عنوان شاخص اصلی آسیب ناشی از ROS به غشای سلولی است با استفاده از سنجش مقادیر میزان مالون دی آلدئید (MDA) به عنوان یکی از محصولات نهایی ناشی از پراکسیداسیون اسیدهای چرب اشباع نشده، قابل بررسی است.
استرس اکسیداتیو عدم تعادل بین مولکولهای اکسیدان و آنتی اکسیدان، به نفع اکسیدانها است که باعث پیری و بیماری میشود. آنتی اکسیدانها یا عوامل ضد اکسیداسیون، با اتصال به عوامل استرس زا، اثر اکسیدانهای خطرناک را کاهش داده و قدرت تخریب آنها را کم میکنند. به همین دلیل سنجش میزان آنتی اکسیدانها، از ارزش بالایی برخوردار است که این ترکیبات در هیستوژنوتک با استفاده از کیتهای اختصاصی مورد ارزیابی قرار میگیرد. به طور مثال سنجش سوپراکسید دیسموتاز (SOD)، کاتالاز (CAT)، گلوتاتیون پراکسیداز (GPX) و همچنین برخی از آنتی اکسیدانها (مانند GSH) به روش غیر آنزیمی و با استفاده از ترکیبات شیمیایی در این مرکز مورد بررسی قرار می گیرد.. آسیبهای ناشی از عوامل استرس زا در سه سطح قابل بررسی است: ۱-آسیب پروتئینی، ۲-آسیب در متابولیسم چربی ۳-آسیب به DNA . محتوای پروتئین کربونیل (PC) یک نشانگر رایج جهت بررسی آسیب در سطح پروتئین است. پراکسیداسیون لیپیدها (اسیدهای چرب)معمولاً به عنوان شاخص اصلی آسیب ناشی از ROS به غشای سلولی است با استفاده از سنجش مقادیر میزان مالون دی آلدئید (MDA) به عنوان یکی از محصولات نهایی ناشی از پراکسیداسیون اسیدهای چرب اشباع نشده، قابل بررسی است.
ncRNA در سالهای اخیر در تعداد زیادی از فرآیندهای بیولوژیکی و در پاتوفیزیولوژی بیماریهای انسانی نقش داشته اند. MiRNA ها و lncRNA ها به عنوان RNA غیر رمز گذار، تنظیم کننده کلیدی شبکه های بیان ژن هستند که فرآیندهای بیولوژیکی متنوعی از جمله تکثیر، تمایز، آپوپتوز، متابولیسم، برهمکنش های پاتوژن را کنترل می کنند. با توجه به اهمیت روزافزون miRNA ها در مکانیسم های بیولوژیکی متعدد، چندین روش مبتنی بر qPCR برای تجزیه و تحلیل بیان miRNA در سال های اخیر توسعه یافته است. طراحی پرایمر برای miRNA RT-qPCR به دلیل طول کوتاه آن (میانگین ۲۲ نوکلئوتید) با چالش بزرگی همراه است . در این مجموعه بررسی بیان micro RNA از طریق طراحی اختصاصی و عمومی Stem loop و همچنین افزایش طول micro RNA (میکرو آر ان ای) با استفاده از روش آنزیمی Poly A Polymerase صورت می گیرد.
فرآیند کلونینگ به منظور انتقال ژن، توسط یک حامل (وکتور بیانی) به سلول میزبان و به دنبال آن تکثیر و افزایش بیان پروتئین می باشد. اهمیت فرآیند کلونینگ در دستیابی به حجم بالایی از یک نوع پروتئین خاص به صورت خالص می باشد. به همین منظور در صنعت داروسازی و تولید پروتئین های نوترکیب بسیار حائز اهمیت است. علاوه بر این دانشمندان با استفاده از توسعه روزافزون استراتژی های شبیه سازی مولکولی توانسته اند فیزیولوژی، مکانیسم های مولکولی و الگوهای بیان ژن را درسلول ها و ارگانیسم ها درک کنند. جهت طراحی وکتور مناسب به عنوان ناقل یک ژن در سلول میزبان، لازم است قطعه DNA مورد نظر طوری طراحی گردد که در دو انتهای آن جایگاه برش، برای آنزیم های برشگر مورد نظر وجود داشته باشد. در هیستوژنوتک طراحی وکتور بیانی با استفاده از نرم افزارهای اختصاصی (Snapgene) برای کنترل جایگاه های برش، توالی ژنوم و ویژگی های مربوط به وکتور صورت گرفته و فرآیند کلونینگ و غربالگری کلنی های های نوترکیب از طریق رنگ آمیزی و در نهایت PCR صورت می گیرد.
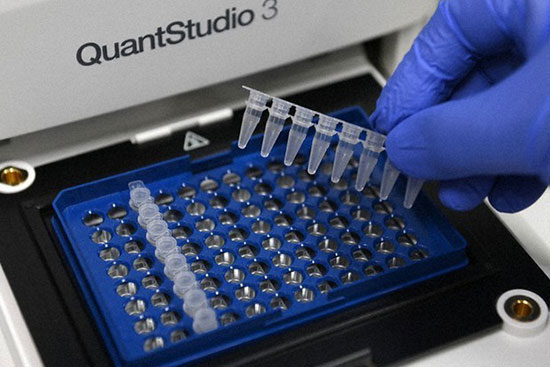
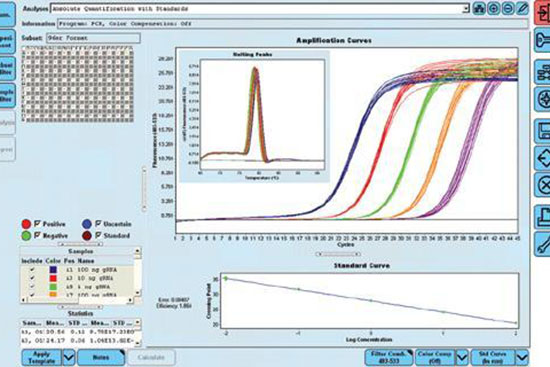
روش ریل تایم پی سی آر، از ادغام PCR کلاسیک با ویژگی های شیمی – فیزیک مواد فلورسنت ابداع شده است که از دستگاه دیجیتال برای اندازه گیری میزان تغییرات فلورسنت در نمونه های مورد سنجش استفاده می شود. این روش به صورت کمّی توانایی اندازه گیری کپی های الگو (cDNA, DNA) را دارد. qPCR برای تشخیص سریع انواع سرطان، ناهنجاری های ژنتیکی، بیماریهای عفونی مانند انواع جدید آنفولانزا و کروناویروس به صورت عمده در آزمایش های تشخیصی استفاده می شود. در هیستوژنوتک بررسی بیان انواع ژن ها با استفاده از دستگاه ترموسایکلرABI مدل Step one و کیت های تک مرحله ای و یا چند مرحله ای به صورت پیوسته در طرح های پژوهشی مختلف مورد استفاده قرار گرفته و نتایج حاصل از آن پس از نرمال سازی با ژن رفرنس به صورت نسبی (fold change) گزارش می گردد. (ریل تایم پی سی آر)
مولتی پلکس پی سی آر یک نوع واکنشPCR است که در آن دو یا چند جایگاه هدف در یک واکنش PCR مورد ارزیابی قرار می گیرد. برای این منظور پرایمر های اختصاصی هر توالی با سایزهای مختلف طراحی و از طریق ژل الکتروفورز تفکیک خواهند شد. در این روش می توان دو یا چند عامل با زمان و هزینه کم تشخیص داده شوند. علاوه بر این در موارد کمبود نمونه DNA نیز می توان از این روش استفاده نمود. طراحی پرایمر برای این نوع از PCR بسیار حائز اهمیت بوده که در هیستوژنوتک با استفاده از نرم افزارهای رایج در این زمینه (PrimerPlex و … ) استفاده می گردد. با استفاده از این نرم افزارهای طراحی پرایمر، عدم تطابق Tm پرایمها بررسی و به حداقل رسانده می شود. در این مجموعه از مولتی پلکس PCR در طرح های پژوهشی مختلف از جمله در ژنتیک میکروبی، شناسایی پاتوژن و تشخیص میکروب ها، تشخیص درصد خلوص محصولات دامی استفاده می شود.
بررسی تغییرات اپی ژنتیکی بر خلاف تغییرات ژنیِ قابل توارث، فاقد تغییر در توالی DNA می باشد. اگرچه تقریباً همه سلولهای موجودات زنده حاوی اطلاعات ژنتیکی یکسانی هستند، اما همه ژنها به طور همزمان بیان نمی شوند. متیلاسیون DNA توسط خانواده ای از متیل ترانسفرازهای DNA (Dnmts) کاتالیز می گردد. تحقیقات ژنی مربوط به متیلاسیون DNA، می تواند اطلاعات مهمی در مورد فرآیندهای اساسی ارائه دهد که به طور معمول به تعیین سرنوشت و عملکرد سلول ها کمک می کند. یکی از متداول ترین روشها برای تعیین وضعیت متیلاسیون در توالی DNA تیمار DNA با بیسولفیت است که باعث می شود بازهای غیر متیله سیتوزین گروه آمین خود را از دست بدهد و به باز یوراسیل تبدیل شود. هیستوژنوتک با استفاده تجربه های متفاوت در این زمینه، بررسی متیلاسیون را از دو روش MSP ((methylation-Specific PCR و BSP (bisulfite Sequencing PCR)، با استفاده از پرایمر های اختصاصی انجام می دهد.
وقوع همزمان دو یا چند ژنوتیپ یا آلل ناپیوسته در یک جمعیت را پلی مورفیسم ژنتیکی می گویند. مطالعه پلی مورفیسم ژنی زمینه جدیدی را برای بررسی پاتوژنز و پیشرفت بیماریها و ماهیت تنوع فنوتیپی ایجاد می کند که در آن علاوه بر علل ژنتیکی اختلالات، می توان با تعیین تغییرات DNA، استعداد ابتلا به بیماری یا گزینه های درمانی را مشخص نمود. طیف گسترده ای از روش ها برای تشخیص جهش ها استفاده می شوند که نوع نوکلئیک اسید (DNA یا RNA)، نوع نمونه (خون ، بافتها یا غیره) و تعداد جهش ها در انتخاب روش موثر است. در هیستوژنوتک بواسطه RFLP، جهش های نقطه ای را که منجر به تغییر توالی و محل اثر برش توسط اندونوکلئازهای محدود الاثر می شود، با استفاده از آنزیم های برشگر اختصاصی مشخص می شود. علاوه بر این با استفاده از تکنیک ARMS-PCR جهش های نقطه ای، ژنوتیپ (حالت عادی ، هتروزیگوت و هموزیگوت) یک نمونه قابل تعیین است. در مواردی که که نیاز به تحلیل کلی از یک بخش کوچک از توالی ژنوم باشد، از توالی یابی سانگر استفاده خواهد شد.
تلومرها آرایه های پشت سر هم و تکراری TTAGGGn در انتهای همه کروموزوم های خطی هستند که نقش حفاظتی از انتهای کروموزوم را با تشکیل ساختارهای تلومری مخصوص به نام حلقه های T ایفا می کنند. در بسیاری از انواع سلولهای سوماتیک انسان فعالیت تلومراز کمی وجود دارد که این امر منجر به از بین رفتن تلومر می شود .همچنین کوتاه شدن تدریجی تلومر می تواند منجر به پیری و اختلالات ژنتیکی موثر بر تلومرها (تلومروپاتی) مانند بیماریهای مرتبط با سن مانند ناباروری، آرتریت، دیابت، سرطان، بیماریهای قلبی عروقی و عصبی می گردد. شناسایی آن در آسیب شناسی های ژنتیکی مربوط به سن در انسان کمک کند. در هیستوژنوتک بررسی طول تلومر، علاوه بر سنجش میزان بیان پروتئین های موثر در این زمینه (TERT-TRF) ) از طریق سنجش آن در سطح DNA با استفاده از روش PCR کمّی صورت می گیرد. در این روش تعداد کپی های تکراری تلومر (T) به تعداد نسخه تک کپی ژن نسبت (نسبت T/S) محاسبه می شود.
کروماتین به عنوان الگوی فیزیولوژیکی اطلاعات ژنتیکی یوکاریوتها، با طیف متنوعی از تغییرات پس از ترجمه همراه است. هیستونها به عنوان مولکولهای سازندهی نوکلئوزوم به شمار میروند که یک ساختار اکتامری را برای بستهبندی DNA در یوکاریوتها فراهم میکند. واریانت هیستونی، نقش کلیدی در متنوع کردن ساختار کروماتین دارند. پژوهشگران معتقدند بسیاری از بیماری های چند فاکتوری و شایع مانند دیابت، آلزایمر و … تنها از طریق توارث ایجاد نمی شوند و این تغییرات محیطی (اپیژنتیکی) در سطح DNA و هیستونها است که می تواند بیان ژنهای درگیر در این اختلالات را متاًثر سازد. تغییرات پس از ترجمه انتهای آمینی هیستونها در یکجا میتواند باعث رونویسی و در جای دیگر مانع از آن شود. عوامل محیطی متعددی میتوانند تأثیر خود را با ایجاد تغییرات استیلاسیونی، فسفریلاسیونی و متیلاسیونی در هیستونها اعمال نمایند.. در هیستوژنوتک بررسی تغییرات اپیژنتیکی برای استیلاسیون، متیلاسیون و شناسایی انواع واریانت های هیستونی، با بررسی بیان کمی ژنهای اختصاصی در این زمینه صورت می گیرد.
بررسی Noncoding RNA (lncRNA و microRNA)
ncRNA در سالهای اخیر در تعداد زیادی از فرآیندهای بیولوژیکی و در پاتوفیزیولوژی بیماریهای انسانی نقش داشته اند. MiRNA ها و lncRNA ها به عنوان RNA غیر رمز گذار، تنظیم کننده کلیدی شبکه های بیان ژن هستند که فرآیندهای بیولوژیکی متنوعی از جمله تکثیر، تمایز، آپوپتوز، متابولیسم، برهمکنش های پاتوژن را کنترل می کنند. با توجه به اهمیت روزافزون miRNA ها در مکانیسم های بیولوژیکی متعدد، چندین روش مبتنی بر qPCR برای تجزیه و تحلیل بیان miRNA در سال های اخیر توسعه یافته است. طراحی پرایمر برای miRNA RT-qPCR به دلیل طول کوتاه آن (میانگین ۲۲ نوکلئوتید) با چالش بزرگی همراه است . در این مجموعه بررسی بیان micro RNA از طریق طراحی اختصاصی و عمومی Stem loop و همچنین افزایش طول micro RNA (میکرو آر ان ای) با استفاده از روش آنزیمی Poly A Polymerase صورت می گیرد.
طراحی وکتورهای بیانی
فرآیند کلونینگ به منظور انتقال ژن، توسط یک حامل (وکتور بیانی) به سلول میزبان و به دنبال آن تکثیر و افزایش بیان پروتئین می باشد. اهمیت فرآیند کلونینگ در دستیابی به حجم بالایی از یک نوع پروتئین خاص به صورت خالص می باشد. به همین منظور در صنعت داروسازی و تولید پروتئین های نوترکیب بسیار حائز اهمیت است. علاوه بر این دانشمندان با استفاده از توسعه روزافزون استراتژی های شبیه سازی مولکولی توانسته اند فیزیولوژی، مکانیسم های مولکولی و الگوهای بیان ژن را درسلول ها و ارگانیسم ها درک کنند. جهت طراحی وکتور مناسب به عنوان ناقل یک ژن در سلول میزبان، لازم است قطعه DNA مورد نظر طوری طراحی گردد که در دو انتهای آن جایگاه برش، برای آنزیم های برشگر مورد نظر وجود داشته باشد. در هیستوژنوتک طراحی وکتور بیانی با استفاده از نرم افزارهای اختصاصی (Snapgene) برای کنترل جایگاه های برش، توالی ژنوم و ویژگی های مربوط به وکتور صورت گرفته و فرآیند کلونینگ و غربالگری کلنی های های نوترکیب از طریق رنگ آمیزی و در نهایت PCR صورت می گیرد.
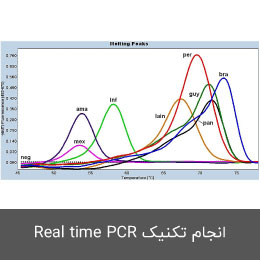
انجام تکنیک Real time PCR (ریل تایم پی سی آر)
روش ریل تایم پی سی آر، از ادغام PCR کلاسیک با ویژگی های شیمی – فیزیک مواد فلورسنت ابداع شده است که از دستگاه دیجیتال برای اندازه گیری میزان تغییرات فلورسنت در نمونه های مورد سنجش استفاده می شود. این روش به صورت کمّی توانایی اندازه گیری کپی های الگو (cDNA, DNA) را دارد. qPCR برای تشخیص سریع انواع سرطان، ناهنجاری های ژنتیکی، بیماریهای عفونی مانند انواع جدید آنفولانزا و کروناویروس به صورت عمده در آزمایش های تشخیصی استفاده می شود. در هیستوژنوتک بررسی بیان انواع ژن ها با استفاده از دستگاه ترموسایکلرABI مدل Step one و کیت های تک مرحله ای و یا چند مرحله ای به صورت پیوسته در طرح های پژوهشی مختلف مورد استفاده قرار گرفته و نتایج حاصل از آن پس از نرمال سازی با ژن رفرنس به صورت نسبی (fold change) گزارش می گردد. (ریل تایم پی سی آر)
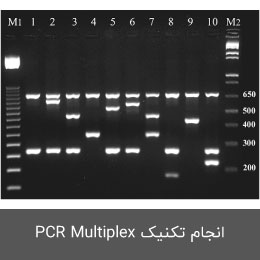
انجام تکنیک PCR Multiplex
مولتی پلکس پی سی آر یک نوع واکنشPCR است که در آن دو یا چند جایگاه هدف در یک واکنش PCR مورد ارزیابی قرار می گیرد. برای این منظور پرایمر های اختصاصی هر توالی با سایزهای مختلف طراحی و از طریق ژل الکتروفورز تفکیک خواهند شد. در این روش می توان دو یا چند عامل با زمان و هزینه کم تشخیص داده شوند. علاوه بر این در موارد کمبود نمونه DNA نیز می توان از این روش استفاده نمود. طراحی پرایمر برای این نوع از PCR بسیار حائز اهمیت بوده که در هیستوژنوتک با استفاده از نرم افزارهای رایج در این زمینه (PrimerPlex و … ) استفاده می گردد. با استفاده از این نرم افزارهای طراحی پرایمر، عدم تطابق Tm پرایمها بررسی و به حداقل رسانده می شود. در این مجموعه از مولتی پلکس PCR در طرح های پژوهشی مختلف از جمله در ژنتیک میکروبی، شناسایی پاتوژن و تشخیص میکروب ها، تشخیص درصد خلوص محصولات دامی استفاده می شود.
بررسی متیلاسیون در ژنوم به روش BSP و MSP
بررسی تغییرات اپی ژنتیکی بر خلاف تغییرات ژنیِ قابل توارث، فاقد تغییر در توالی DNA می باشد. اگرچه تقریباً همه سلولهای موجودات زنده حاوی اطلاعات ژنتیکی یکسانی هستند، اما همه ژنها به طور همزمان بیان نمی شوند. متیلاسیون DNA توسط خانواده ای از متیل ترانسفرازهای DNA (Dnmts) کاتالیز می گردد. تحقیقات ژنی مربوط به متیلاسیون DNA، می تواند اطلاعات مهمی در مورد فرآیندهای اساسی ارائه دهد که به طور معمول به تعیین سرنوشت و عملکرد سلول ها کمک می کند. یکی از متداول ترین روشها برای تعیین وضعیت متیلاسیون در توالی DNA تیمار DNA با بیسولفیت است که باعث می شود بازهای غیر متیله سیتوزین گروه آمین خود را از دست بدهد و به باز یوراسیل تبدیل شود. هیستوژنوتک با استفاده تجربه های متفاوت در این زمینه، بررسی متیلاسیون را از دو روش MSP ((methylation-Specific PCR و BSP (bisulfite Sequencing PCR)، با استفاده از پرایمر های اختصاصی انجام می دهد.
بررسی پلیمورفیسم و جهش ژنی به روش RFLP، Nested PCR،ARMS وتوالی یابی ژنوم
وقوع همزمان دو یا چند ژنوتیپ یا آلل ناپیوسته در یک جمعیت را پلی مورفیسم ژنتیکی می گویند. مطالعه پلی مورفیسم ژنی زمینه جدیدی را برای بررسی پاتوژنز و پیشرفت بیماریها و ماهیت تنوع فنوتیپی ایجاد می کند که در آن علاوه بر علل ژنتیکی اختلالات، می توان با تعیین تغییرات DNA، استعداد ابتلا به بیماری یا گزینه های درمانی را مشخص نمود. طیف گسترده ای از روش ها برای تشخیص جهش ها استفاده می شوند که نوع نوکلئیک اسید (DNA یا RNA)، نوع نمونه (خون ، بافتها یا غیره) و تعداد جهش ها در انتخاب روش موثر است. در هیستوژنوتک بواسطه RFLP، جهش های نقطه ای را که منجر به تغییر توالی و محل اثر برش توسط اندونوکلئازهای محدود الاثر می شود، با استفاده از آنزیم های برشگر اختصاصی مشخص می شود. علاوه بر این با استفاده از تکنیک ARMS-PCR جهش های نقطه ای، ژنوتیپ (حالت عادی ، هتروزیگوت و هموزیگوت) یک نمونه قابل تعیین است. در مواردی که که نیاز به تحلیل کلی از یک بخش کوچک از توالی ژنوم باشد، از توالی یابی سانگر استفاده خواهد شد.


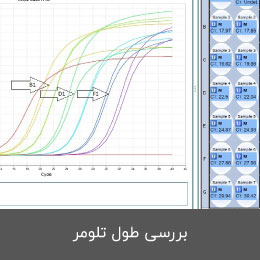
بررسی طول تلومر با استفاده از روش PCR کمّی
تلومرها آرایه های پشت سر هم و تکراری TTAGGGn در انتهای همه کروموزوم های خطی هستند که نقش حفاظتی از انتهای کروموزوم را با تشکیل ساختارهای تلومری مخصوص به نام حلقه های T ایفا می کنند. در بسیاری از انواع سلولهای سوماتیک انسان فعالیت تلومراز کمی وجود دارد که این امر منجر به از بین رفتن تلومر می شود .همچنین کوتاه شدن تدریجی تلومر می تواند منجر به پیری و اختلالات ژنتیکی موثر بر تلومرها (تلومروپاتی) مانند بیماریهای مرتبط با سن مانند ناباروری، آرتریت، دیابت، سرطان، بیماریهای قلبی عروقی و عصبی می گردد. شناسایی آن در آسیب شناسی های ژنتیکی مربوط به سن در انسان کمک کند. در هیستوژنوتک بررسی طول تلومر، علاوه بر سنجش میزان بیان پروتئین های موثر در این زمینه (TERT-TRF) ) از طریق سنجش آن در سطح DNA با استفاده از روش PCR کمّی صورت می گیرد. در این روش تعداد کپی های تکراری تلومر (T) به تعداد نسخه تک کپی ژن نسبت (نسبت T/S) محاسبه می شود.
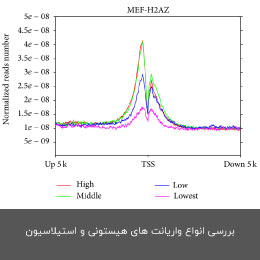
بررسی انواع واریانت های هیستونی و استیلاسیون
کروماتین به عنوان الگوی فیزیولوژیکی اطلاعات ژنتیکی یوکاریوتها، با طیف متنوعی از تغییرات پس از ترجمه همراه است. هیستونها به عنوان مولکولهای سازندهی نوکلئوزوم به شمار میروند که یک ساختار اکتامری را برای بستهبندی DNA در یوکاریوتها فراهم میکند. واریانت هیستونی، نقش کلیدی در متنوع کردن ساختار کروماتین دارند. پژوهشگران معتقدند بسیاری از بیماری های چند فاکتوری و شایع مانند دیابت، آلزایمر و … تنها از طریق توارث ایجاد نمی شوند و این تغییرات محیطی (اپیژنتیکی) در سطح DNA و هیستونها است که می تواند بیان ژنهای درگیر در این اختلالات را متاًثر سازد. تغییرات پس از ترجمه انتهای آمینی هیستونها در یکجا میتواند باعث رونویسی و در جای دیگر مانع از آن شود. عوامل محیطی متعددی میتوانند تأثیر خود را با ایجاد تغییرات استیلاسیونی، فسفریلاسیونی و متیلاسیونی در هیستونها اعمال نمایند.. در هیستوژنوتک بررسی تغییرات اپیژنتیکی برای استیلاسیون، متیلاسیون و شناسایی انواع واریانت های هیستونی، با بررسی بیان کمی ژنهای اختصاصی در این زمینه صورت می گیرد.
یکی از اشتباهات رایج بین دانشجویان، به کار بردن واژه RT-PCR برای سه تست کاملا متفاوت ,cDNA Synthesis Real Time PCR , RT-PCR است. در صورتی که این سه تست دارای تفاوت هایی هستند. اصطلاح RT-PCR برای هر سه مورد سنتز cDNA qPCR, و یا pcr کلاسیک استفاده می شود. در سنتز cDNA از RNA به عنوان الگو استفاده می شود که آنزیم رونوشت بردار معکوس در یک چرخه دمایی با استفاده از توالی پرایمر (الیگو dt و یا رندوم هگزامر) از روی DNA می سازد. در صورتی که در PCR کلاسیک و qPCR ازcDNA به عنوان الگو استفاده شده و در چندین سیکل دمایی از روی یک ژن با پرایمر اختصاصی تکثیر خواهد شد. تفاوت این دو PCR در این است که در qPCR از ترموسایکلر ویژه استفاده می شود که می تواند سیگنال فلورسنت بازتاب شده را تشخیص داده و در نهایت نتایج را به صورت کمّی گزارش کند. اما در PCR کلاسیک نتایج به صورت نیمه کمّی گزارش می شود که در آن محصولات PCR پس از تفکیک در ژل الکتروفورز با نرم افزار اختصاصی به صورت کمّی قابل گزارش است.
فریزرهای با دمای منفی ۸۰ درجه سانتی گراد یک گزینه مناسب برای ذخیره سازی طولانی مدت مواد بیولوژیکی است. این دما از تجزیه اسیدهای نوکلئیک، پروتئین ها و بسیاری از مولکول های بیولوژیکی جلوگیری می کند. در مواردی که دمای منفی ۸۰ موجود نباشد و یا شرایط ارسال نمونه با یخ خشک میسر نیست، نمونه ها را می توان در محلول های نمک سولفات آبی غوطه ور نمود. مشابه تجاری و آماده این ترکیب با نام RNALater موجود می باشد که می توان بر روی بافت های جدا شده از جاندار و یا سلول اضافه نمود. بافت های جامد را می توان به مدت یک هفته در RNALater در دمای اتاق ذخیره کرد. برای نگهداری طولانی مدت لازم است نمونه های حاوی این محلول در منفی ۲۰ ذخیره گردد. برای جابجایی و انتقال نمونه های فریز شده، لازم است دمای منفی ۸۰ را با استفاده از یخ خشک صنعتی حفظ شده و به سرعت نمونه ها به آزمایشگاه مورد نظر انتقال داده شوند.
در بررسی متیلاسیون، اساس عملکرد تکنیکهایی که پایهی آنها تغییرات بیوشیمیایی توسط بیسولفیت است به این صورت می باشد که سیتوزینهای غیرمتیله به یوراسیل تبدیل میشوند ولی بر روی سیتوزینهای متیله عملکردی ندارند. بنابراین بی سولفیت بسته به حضور یا عدم حضور متیل روی سیتوزین تغییرات ویژهای را در DNA ایجاد میکند که با روشهای PCR، توالییابی و MicroRNA قابل ارزیابی است. امروزه چندین روش برای بررسی آنالیز متیلاسیون DNA وجود دارد. با این حال، هیچ روش خاصی به عنوان تکنیک “اصلی” شناخته نشده است. هر روش دارای مزایا و معایب است. روشهایی که در آن میتوان از کیتهای تجاری برای تشخیص متیلاسیون استفاده کرد به عنوان بهترین روش برای تیمار بیسولفیت شناخته می شوند.
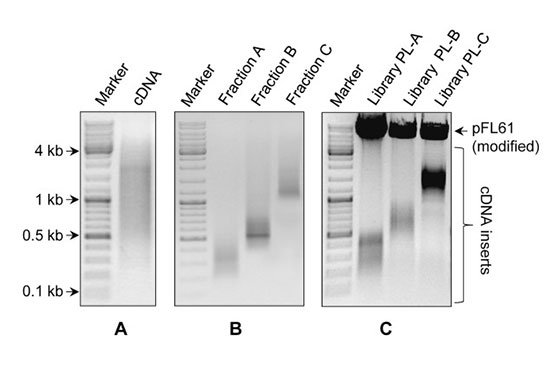
بسته به نوع تکنیک، کیفیت مورد نیاز برای RNA و DNA استخراج شده متفاوت است. در انواع روش های توالی یابی مانند NGS به کیفیت بالایی از RNA نیاز است در صورتی که برای فرآیند ریل تایم پی سی آر نمونه با کیفیت متوسط هم کافی است. به همین منظور لازم است از کیفیت نمونه استخراج شده قبل از انجام کار مطلع شد. در بررسی اولیه نمونه استخراج شده نمونه را با استفاده ژل آگاروز ۱.۵% تفکیک می کنند. در یک نمونه با کیفیت از DNA باند مربوط به آن در بالای ژل مشاهده می شود. در نمونه های RNA، ۳ قطعه RNA , 18 S,28 S, 5.8 S به صورت ۳ باند جداگانه تفکیک می گردد. وجود اسمیر در ژل نشان از تخریب RNA می باشد. در یک روش دیگر برای تعیین دقیق کیفیت RNAاستخراج شده از دستگاه Bioanalyzer استفاده می شود که با استفاده از آن عنوان عدد یکپارچگی RNA (RIN Value) نمایش داده می شود. در نمونه های تخریب شدهRNA ، ارتفاع قله برای قله های rRNA 28S و ۱۸S کاهش می یابد و در نمونه های با کیفیت، قله های به صورت تفکیک شده از هم مشاهده خواهد شد. هر چقدر عدد RNA به ۱۰ نزدیکتر باشد، آن نمونه از کیفیت بالاتری برخوردار است.
انتخاب روش PCR به طراحی مطالعه ارتباط دارد. به طور مثال برای بررسی بیان ژن در سطح mRNAو non Coding RNA از روش هایPCR کمّی و یا PCR کلاسیک می توان استفاده نمود. بررسی تغییرات ژنومی، جهش های ژنی و پلی مورفیسم از DNA استفاده می شود و انتخاب نوع PCR بسته به نوع نمونه، روش مطالعه و حساسیت مطالعه، اطلاعات موجود در بانک ژنی و… قابل تعیین است. از روش ARMS برای شناسایی جهش های نقطه ای استفاده می شود که در آن پرایمر بر اساس آلل های مختلف به صورت انحصاری طراحی می شود. از RFLP برای تشخیص انواع پلی مورفیسم، ژنوتایپ و غربالگری جهش های شناسایی شده استفاده می گردد که اساس آن بر تفاوت بین سایز قطعات در برش های آنزیمی محدودالاثر محصولات حاصل از PCR است. از روش Nested برای ردیابی توالی DNA هدف استفاده می گردد که در آن از دو جفت پرایمر تو در تو استفاده می شود. اختصاصیت این روش به دلیل نوع پرایمر بسیار بالا می باشد. علاوه بر این می توان از انواع pcr برای شناسایی انواع سویه های میکروبی، انگل و .. استفاده نمود به طور مثال multiplex pcr قابلیت شناسایی همزمان چند سویه باکتری و یا انگل را در یک واکنش دارد. در این تست از چندین جفت پرایمر اختصاصی به صورت همزمان استفاده می گردد.
نرمافزارهای متعددی همچون ,UCSC ,Primer3 ,MEDUSA,Oligo7 برای طراحی پرایمر وجود دارد که روند طراحی پرایمر را برای کاربر آسان نموده است. پرایمرها را باید پس از طراحی در نرم افزار آنلاین Primer3، در صفحهی Primer BLAST موجود در سایت NCBI((http://www.ncbi.nlm..nih.gov/tools/primer-blast بررسی نموده تا از عدم اتصال آن به سایر ژنها مطمئن شویم. به طور کلی در طراحی پرایمرها باید نکات بسیار مهم و کاربردی زیر را رعایت کرد. – طول آن بین ۱۸ تا ۳۰ نوکلئوتید باشد، – بهتر است محتوایGC بین %۵۰ تا %۶۰ باشد ( افزایش مقدار محتوای GC منجربه افزایش درجه حرارت اتصال و متعاقب آن افزایش استحکام هیبرید و دایمر پرایمر خواهد شد) – عدم وجود تفاوت چشمگیر بین دمای اتصال دو پرایمر پیشرو و پسرو ( در شرایط مطلوب این اختلاف بین منفی ۵ و ۵ باشد) – عدم وجود نواحی مکمل در یک پرایمر و یا پرایمرهای مختلف به خصوص در انتهای ۳پرین (برای ایجاد اتصال محکم لازم است انتهای ۳ یکی از نوکلئوتیدهای G یاC باشد) – Tm پرایمرها ترجیحاً بین۶۰-۵۸ درجه سانتیگراد طراحی شود و حداکثر اختلاف بینTm دو پرایمر ۳ درجه باشد – طراحی پرایمر از نواحی پایدار (احتمال کمتر وقوع جهش) DNA هدف صورت گیرد – برای پیشگیری از تکثیر DNA ژنومی باید پرایمرها طوری انتخاب شود که سر ۳پرین آن روی یک اگزون و سر ۵پرین آن روی اگزون دیگر باشد به این ترتیب چون تکثیر از cDNA انجام میشود که فاقد اینترون است،DNA ژنومی تکثیر نخواهد شد.
ژن رفرنس (housekeeping gene) برای اندازه گیری بیان نسبی یک ژن بدون نیاز به محلول استاندارد مورد استفاده قرار می گیرد. یک ژن رفرنس مناسب باید بیان پایداری در بین نمونه ها داشته باشد و تحت تاثیر تغییرات فیزیولوژیکی مختلف قرار نگیرید. ژن رفرنس معمولاً برای نرمال سازی داده های RT-qPCR انتخاب می شوند. از آنجا که این ژنها در متابولیسم پایه سلولی دخیل هستند، فرض بر این است که آنها به طور اساسی بیان شده و تحت تاثیر تیمارهای مختلف قرار نمی گیرند. با این حال، ژن های رفرنس ممکن است با توجه به تغییر شرایط فیزیولوژیک و نوع بافت تغییر کنند به همین دلیل برای هر مطالعه لازم است بسته به نوع بافت و نوع مطالعه، ژن رفرنس را انتخاب نمود. علاوه بر این توصیه می شود از حداقل دو ژن مرجع برای اطمینان از نرمال سازی نتایج بیان ژن استفاده شود. یک ژن رفرنس در حالت ایده آل باید پایدار بوده و در سلولها و بافتهای مورد نظر که در شرایط عادی یا حالت بیماری تغییراتی را نشان نمی دهد استفاده شود. از ژن های رفرنس که در مطالعات مختلف استفاده می شود می توان به ۱۸sRNA,GAPDH, β-Actin,B2M,.. اشاره کرد.
یکی از مهمترین کاربردهای Real-Time PCR تعیین کمّی مقدار ژن بیان شده است که به دو روش مطلق و نسبی صورت میگیرد. روش تعیین کمیت نسبی به منظور تعیین نسب تعداد نسخههای mRNA و بیان ژن مورد نظر است. در این روش تعداد مهم نبوده و فقط کاهش و یا افزایش بیان ژن مد نظر می باشد. که این افزایش و یا کاهش را با یک ژن استاندارد یا مرجع مقایسه میکنند. اساس کمیتسنجی نسبی بر پایهی تعیین نسبت بیان ژن مورد نظر به ژن مرجع است. در این روش بازده PCR هر دو ژن باید تقریباً برابر باشد، زیرا در غیر این صورت نتیجهی آزمایش با خطای بالایی همراه است. سپس تفاوت بین CT هر دو ژن رفرنس و مرجع صورت گیرد) (۲-∆CT در این حالت نرمال سازی نتایج بر اساس ژن رفرنس محاسبه می گردد. اگر در گزارش نتایج PCR، بخواهیم تفاوت در میزان بیان را نسبت به یک گروه (معمولا گروه کنترل) نشان دهیم لازم است تفاوت DCT بین گروه های کنترل با سایر گروه ها صورت گیرد(۲-∆∆CT) که در آن گروه کنترل عدد۱ را خواهد داشت و سایر گروه ها عدد بالاتر و یا پایین تر از ۱ را نشان می دهند که نشان از افزایش و یا کاهش بیان ژن مورد نظر می باشد.
![]()
اگر بازده PCR را کامل در نظر بگیریم فرمول زیر قابل استفاده است:
![]()
با استفاده از نتایج خوانش در نانودراپ در طول موج ۲۶۰- ۲۸۰ نانومتر می توان غلظت و کیفیت RNA را گزارش نمود. در این نتایج نسبتOD۲۶۰/OD۲۸۰ بیانگر میزان خلوص RNA استخراج شده میباشد و رابطهی مستقیم با آلودگی RNA به پروتئین دارد. بنابراین نزدیک بودن این نسبت به عدد۲ نشان دهندهی عدم آلودگی به پروتئین است. از نسبتOD۲۶۰/OD۲۳۰ نیز برای بررسی میزان آلودگی RNA، به مواد بکار برده شده برای استخراج (فنول و ترکیبات نمکی) استفاده میشود. هر چقدر مقدار آن به عدد۲ نزدیکتر باشد شرایط را برای انجام آزمایشات مطلوبتر خواهد کرد.
برای برطرف کردن آلودگی نمکی (میزان OD۲۶۰/OD۲۳۰ <1.8)، بهترین روش شستشوی RNA می باشد. اگر RNA استخراج شده با استفاده از ترایزول (روش فنل کلروفرم) باشد، بهتر است آن را با اتانول شستشو داده تا نمک زدایی صورت گیرد. در روش استفاده از ستون و کیت، با افزایش تعداد دفعات شستشو با اتانول ۷۰-۸۰ درصد میزان نمک ها را از ستون می توان از بین برد. برای حذف فنل بالا در نمونه RNA می توان RNA خود را در Tris-EDTA حل کرده و میزان یک حجم از کلروفورم به آن اضافه نمود و سپس سانترفیوژ کرد.
وجود قلل متعدد و یا باندهای اضافه در نتایج PCR می تواند دلایل مختلفی را به همراه داشته باشد. به طور کلی وجود هر پیک و یا باند نشان از یک محصول است که در فرآیند PCR تکثیر یافته است. به طور کلی محصولات با دمای ذوب پایین تر معمولا نشان دهنده دایمر پرایمر و یا محصولات اختصاصی با طول کم می باشند. در ژل الکتروفورز این محصولات در قسمت پایین تر ژل قرار می گیرند. برای حذف این محصولات باید پرایمر را به صورت اختصاصی تر طراحی نمود و از وجود اتصالات میان جفت پرایمر و تشکیل دایمر مطمئن شد. در بعضی موارد با تغییر دمای Anneling یا دمای اتصال پرایمر می توان این محصول را حذف نمود. در مواردی که Tm محصولات اضافه pcr بالا باشد، ممکن است توالی پرایمر، یک قطعه دیگر در ژنوم را شناسایی نموده و یا اینکه نمونه به یک ارگانیسم دیگر آلوده باشد. در این حالت علاوه بر اینکه باید دقت شود نمونه ها آلودگی با ارگانیسم دیگر نداشته باشند می توان پرایمر را تعویض نمود و یا در بعضی موارد با تغییر دما تکثیر این محصول را مهار کرد. برای اینکه بفهمیم کدام محصول ما (کدام قله( اختصاصی است، باید محصولات را با استفاده از ژل الکتروفورز تفکیک کرده و در نهایت با توجه به سایز قطعه مورد نظر که پرایمر اختصاصی آن طراحی شده، محصول و یا Tm اختصاصی مربوط به ژن هدف را مشخص نمود.
در موارد کاهش غلظت RNA لازم است میزان رسوب RNA را با استفاده از ترکیبات دیگر که روند رسوب آن را بالا می برد افزایش داد. برای انجام این فرآیند در مرحله رسوب RNA با استفاده از الکل، بهتر است از الکل ایزوپروپانول غلیظ استفاده شود . در این مرحله علاوه بر ایزوپروپونال، ترکیباتی از کربوهیدرات های شاخه دار می تواند در به دام انداختن RNA و افزایش رسوب کمک کند. علاوه بر این کاهش دما، می تواند منجر به افزایش رسوب RNA شود. به همین منظور بهتر است پروسه انکوباسیون در ۴ درجه سانتی گراد را پس از اضافه کردن ایزوپروپانول طولانی تر نمود.
فریزرهای با دمای منفی ۸۰ درجه سانتی گراد یک گزینه مناسب برای ذخیره سازی طولانی مدت مواد بیولوژیکی است. این دما از تجزیه اسیدهای نوکلئیک، پروتئین ها و بسیاری از مولکول های بیولوژیکی جلوگیری می کند. در مواردی که دمای منفی ۸۰ موجود نباشد و یا شرایط ارسال نمونه با یخ خشک میسر نیست، نمونه ها را می توان در محلول های نمک سولفات آبی غوطه ور نمود. مشابه تجاری و آماده این ترکیب با نام RNALater موجود می باشد که می توان بر روی بافت های جدا شده از جاندار و یا سلول اضافه نمود. بافت های جامد را می توان به مدت یک هفته در RNALater در دمای اتاق ذخیره کرد. برای نگهداری طولانی مدت لازم است نمونه های حاوی این محلول در منفی ۲۰ ذخیره گردد. برای جابجایی و انتقال نمونه های فریز شده، لازم است دمای منفی ۸۰ را با استفاده از یخ خشک صنعتی حفظ شده و به سرعت نمونه ها به آزمایشگاه مورد نظر انتقال داده شوند.
در بررسی متیلاسیون، اساس عملکرد تکنیکهایی که پایهی آنها تغییرات بیوشیمیایی توسط بیسولفیت است به این صورت می باشد که سیتوزینهای غیرمتیله به یوراسیل تبدیل میشوند ولی بر روی سیتوزینهای متیله عملکردی ندارند. بنابراین بی سولفیت بسته به حضور یا عدم حضور متیل روی سیتوزین تغییرات ویژهای را در DNA ایجاد میکند که با روشهای PCR، توالییابی و MicroRNA قابل ارزیابی است. امروزه چندین روش برای بررسی آنالیز متیلاسیون DNA وجود دارد. با این حال، هیچ روش خاصی به عنوان تکنیک “اصلی” شناخته نشده است. هر روش دارای مزایا و معایب است. روشهایی که در آن میتوان از کیتهای تجاری برای تشخیص متیلاسیون استفاده کرد به عنوان بهترین روش برای تیمار بیسولفیت شناخته می شوند.
بسته به نوع تکنیک، کیفیت مورد نیاز برای RNA و DNA استخراج شده متفاوت است. در انواع روش های توالی یابی مانند NGS به کیفیت بالایی از RNA نیاز است در صورتی که برای فرآیند ریل تایم پی سی آر نمونه با کیفیت متوسط هم کافی است. به همین منظور لازم است از کیفیت نمونه استخراج شده قبل از انجام کار مطلع شد. در بررسی اولیه نمونه استخراج شده نمونه را با استفاده ژل آگاروز ۱.۵% تفکیک می کنند. در یک نمونه با کیفیت از DNA باند مربوط به آن در بالای ژل مشاهده می شود. در نمونه های RNA، ۳ قطعه RNA , 18 S,28 S, 5.8 S به صورت ۳ باند جداگانه تفکیک می گردد. وجود اسمیر در ژل نشان از تخریب RNA می باشد. در یک روش دیگر برای تعیین دقیق کیفیت RNAاستخراج شده از دستگاه Bioanalyzer استفاده می شود که با استفاده از آن عنوان عدد یکپارچگی RNA (RIN Value) نمایش داده می شود. در نمونه های تخریب شدهRNA ، ارتفاع قله برای قله های rRNA 28S و ۱۸S کاهش می یابد و در نمونه های با کیفیت، قله های به صورت تفکیک شده از هم مشاهده خواهد شد. هر چقدر عدد RNA به ۱۰ نزدیکتر باشد، آن نمونه از کیفیت بالاتری برخوردار است.
انتخاب روش PCR به طراحی مطالعه ارتباط دارد. به طور مثال برای بررسی بیان ژن در سطح mRNAو non Coding RNA از روش هایPCR کمّی و یا PCR کلاسیک می توان استفاده نمود. بررسی تغییرات ژنومی، جهش های ژنی و پلی مورفیسم از DNA استفاده می شود و انتخاب نوع PCR بسته به نوع نمونه، روش مطالعه و حساسیت مطالعه، اطلاعات موجود در بانک ژنی و… قابل تعیین است. از روش ARMS برای شناسایی جهش های نقطه ای استفاده می شود که در آن پرایمر بر اساس آلل های مختلف به صورت انحصاری طراحی می شود. از RFLP برای تشخیص انواع پلی مورفیسم، ژنوتایپ و غربالگری جهش های شناسایی شده استفاده می گردد که اساس آن بر تفاوت بین سایز قطعات در برش های آنزیمی محدودالاثر محصولات حاصل از PCR است. از روش Nested برای ردیابی توالی DNA هدف استفاده می گردد که در آن از دو جفت پرایمر تو در تو استفاده می شود. اختصاصیت این روش به دلیل نوع پرایمر بسیار بالا می باشد. علاوه بر این می توان از انواع pcr برای شناسایی انواع سویه های میکروبی، انگل و .. استفاده نمود به طور مثال multiplex pcr قابلیت شناسایی همزمان چند سویه باکتری و یا انگل را در یک واکنش دارد. در این تست از چندین جفت پرایمر اختصاصی به صورت همزمان استفاده می گردد.
نرمافزارهای متعددی همچون ,UCSC ,Primer3 ,MEDUSA,Oligo7 برای طراحی پرایمر وجود دارد که روند طراحی پرایمر را برای کاربر آسان نموده است. پرایمرها را باید پس از طراحی در نرم افزار آنلاین Primer3، در صفحهی Primer BLAST موجود در سایت NCBI((http://www.ncbi.nlm..nih.gov/tools/primer-blast بررسی نموده تا از عدم اتصال آن به سایر ژنها مطمئن شویم. به طور کلی در طراحی پرایمرها باید نکات بسیار مهم و کاربردی زیر را رعایت کرد. – طول آن بین ۱۸ تا ۳۰ نوکلئوتید باشد، – بهتر است محتوایGC بین %۵۰ تا %۶۰ باشد ( افزایش مقدار محتوای GC منجربه افزایش درجه حرارت اتصال و متعاقب آن افزایش استحکام هیبرید و دایمر پرایمر خواهد شد) – عدم وجود تفاوت چشمگیر بین دمای اتصال دو پرایمر پیشرو و پسرو ( در شرایط مطلوب این اختلاف بین منفی ۵ و ۵ باشد) – عدم وجود نواحی مکمل در یک پرایمر و یا پرایمرهای مختلف به خصوص در انتهای ۳پرین (برای ایجاد اتصال محکم لازم است انتهای ۳ یکی از نوکلئوتیدهای G یاC باشد) – Tm پرایمرها ترجیحاً بین۶۰-۵۸ درجه سانتیگراد طراحی شود و حداکثر اختلاف بینTm دو پرایمر ۳ درجه باشد – طراحی پرایمر از نواحی پایدار (احتمال کمتر وقوع جهش) DNA هدف صورت گیرد – برای پیشگیری از تکثیر DNA ژنومی باید پرایمرها طوری انتخاب شود که سر ۳پرین آن روی یک اگزون و سر ۵پرین آن روی اگزون دیگر باشد به این ترتیب چون تکثیر از cDNA انجام میشود که فاقد اینترون است،DNA ژنومی تکثیر نخواهد شد.
یکی از اشتباهات رایج بین دانشجویان ، به کار بردن واژه RT-PCR برای سه تست کاملا متفاوت ,cDNA Synthesis Real Time PCR , RT-PCR است. در صورتی که این سه تست دارای تفاوت های بسیاری هستند. اصطلاح RT-PCR برای هر سه مورد سنتزcDNA qPCR, و یا pcr کلاسیک استفاده می شود. در سنتز cDNA از RNA به عنوان الگو استفاده می شود که آنزیم رونوشت بردار معکوس در یک چرخه دمایی با استفاده از توالی پرایمر(الیگو dt و یا رندوم هگزامر) از روی DNA می سازد. در صورتی که درPCR کلاسیک و qPCR ازcDNA به عنوان الگو استفاده شده و در چندین سیکل دمایی از روی یک ژن با پرایمر اختصاصی تکثیر خواهد شد. تفاوت این دو PCR در این است که در qPCR از ترموسایکلر ویژه استفاده می شود که می تواند سیگنال فلورسنت بازتاب شده را تشخیص داده و در نهایت نتایج را به صورت کمّی گزارش کند. اما در PCR کلاسیک نتایج به صورت نیمه کمّی گزارش می شود که در آن محصولات PCR پس از تفکیک در ژل الکتروفورز با نرم افزار اختصاصی به صورت کمّی قابل گزارش است.
ژن رفرنس (housekeeping gene) برای اندازه گیری بیان نسبی یک ژن بدون نیاز به محلول استاندارد مورد استفاده قرار می گیرد. یک ژن رفرنس مناسب باید بیان پایداری در بین نمونه ها داشته باشد و تحت تاثیر تغییرات فیزیولوژیکی مختلف قرار نگیرید. ژن رفرنس معمولاً برای نرمال سازی داده های RT-qPCR انتخاب می شوند. از آنجا که این ژنها در متابولیسم پایه سلولی دخیل هستند، فرض بر این است که آنها به طور اساسی بیان شده و تحت تاثیر تیمارهای مختلف قرار نمی گیرند. با این حال، ژن های رفرنس ممکن است با توجه به تغییر شرایط فیزیولوژیک و نوع بافت تغییر کنند به همین دلیل برای هر مطالعه لازم است بسته به نوع بافت و نوع مطالعه، ژن رفرنس را انتخاب نمود. علاوه بر این توصیه می شود از حداقل دو ژن مرجع برای اطمینان از نرمال سازی نتایج بیان ژن استفاده شود. یک ژن رفرنس در حالت ایده آل باید پایدار بوده و در سلولها و بافتهای مورد نظر که در شرایط عادی یا حالت بیماری تغییراتی را نشان نمی دهد استفاده شود. از ژن های رفرنس که در مطالعات مختلف استفاده می شود می توان به ۱۸sRNA,GAPDH, β-Actin,B2M,.. اشاره کرد.
یکی از مهمترین کاربردهای Real-Time PCR تعیین کمّی مقدار ژن بیان شده است که به دو روش مطلق و نسبی صورت میگیرد. روش تعیین کمیت نسبی به منظور تعیین نسب تعداد نسخههای mRNA و بیان ژن مورد نظر است. در این روش تعداد مهم نبوده و فقط کاهش و یا افزایش بیان ژن مد نظر می باشد. که این افزایش و یا کاهش را با یک ژن استاندارد یا مرجع مقایسه میکنند. اساس کمیتسنجی نسبی بر پایهی تعیین نسبت بیان ژن مورد نظر به ژن مرجع است. در این روش بازده PCR هر دو ژن باید تقریباً برابر باشد، زیرا در غیر این صورت نتیجهی آزمایش با خطای بالایی همراه است. سپس تفاوت بین CT هر دو ژن رفرنس و مرجع صورت گیرد) (۲-∆CT در این حالت نرمال سازی نتایج بر اساس ژن رفرنس محاسبه می گردد. اگر در گزارش نتایج PCR، بخواهیم تفاوت در میزان بیان را نسبت به یک گروه (معمولا گروه کنترل) نشان دهیم لازم است تفاوت DCT بین گروه های کنترل با سایر گروه ها صورت گیرد(۲-∆∆CT) که در آن گروه کنترل عدد۱ را خواهد داشت و سایر گروه ها عدد بالاتر و یا پایین تر از ۱ را نشان می دهند که نشان از افزایش و یا کاهش بیان ژن مورد نظر می باشد. Ratio=E– {(ΔCTcase) – (ΔCTcontrol)} ΔCT= CTtarget – CTreference اگر بازده PCR را کامل در نظر بگیریم فرمول زیر قابل استفاده است: Ratio=2-{(ΔCTcase) – (ΔCTcontrol)}
با استفاده از نتایج خوانش در نانودراپ در طول موج ۲۶۰- ۲۸۰ نانومتر می توان غلظت و کیفیت RNA را گزارش نمود. در این نتایج نسبتOD۲۶۰/OD۲۸۰ بیانگر میزان خلوص RNA استخراج شده میباشد و رابطهی مستقیم با آلودگی RNA به پروتئین دارد. بنابراین نزدیک بودن این نسبت به عدد۲ نشان دهندهی عدم آلودگی به پروتئین است. از نسبتOD۲۶۰/OD۲۳۰ نیز برای بررسی میزان آلودگی RNA، به مواد بکار برده شده برای استخراج (فنول و ترکیبات نمکی) استفاده میشود. هر چقدر مقدار آن به عدد۲ نزدیکتر باشد شرایط را برای انجام آزمایشات مطلوبتر خواهد کرد.
برای برطرف کردن آلودگی نمکی (میزان OD۲۶۰/OD۲۳۰ <1.8)، بهترین روش شستشوی RNA می باشد. اگر RNA استخراج شده با استفاده از ترایزول (روش فنل کلروفرم) باشد، بهتر است آن را با اتانول شستشو داده تا نمک زدایی صورت گیرد. در روش استفاده از ستون و کیت، با افزایش تعداد دفعات شستشو با اتانول ۷۰-۸۰ درصد میزان نمک ها را از ستون می توان از بین برد. برای حذف فنل بالا در نمونه RNA می توان RNA خود را در Tris-EDTA حل کرده و میزان یک حجم از کلروفورم به آن اضافه نمود و سپس سانترفیوژ کرد.
وجود قلل متعدد و یا باندهای اضافه در نتایج PCR می تواند دلایل مختلفی را به همراه داشته باشد. به طور کلی وجود هر پیک و یا باند نشان از یک محصول است که در فرآیند PCR تکثیر یافته است. به طور کلی محصولات با دمای ذوب پایین تر معمولا نشان دهنده دایمر پرایمر و یا محصولات اختصاصی با طول کم می باشند. در ژل الکتروفورز این محصولات در قسمت پایین تر ژل قرار می گیرند. برای حذف این محصولات باید پرایمر را به صورت اختصاصی تر طراحی نمود و از وجود اتصالات میان جفت پرایمر و تشکیل دایمر مطمئن شد. در بعضی موارد با تغییر دمای Anneling یا دمای اتصال پرایمر می توان این محصول را حذف نمود. در مواردی که Tm محصولات اضافه pcr بالا باشد، ممکن است توالی پرایمر، یک قطعه دیگر در ژنوم را شناسایی نموده و یا اینکه نمونه به یک ارگانیسم دیگر آلوده باشد. در این حالت علاوه بر اینکه باید دقت شود نمونه ها آلودگی با ارگانیسم دیگر نداشته باشند می توان پرایمر را تعویض نمود و یا در بعضی موارد با تغییر دما تکثیر این محصول را مهار کرد. برای اینکه بفهمیم کدام محصول ما (کدام قله( اختصاصی است، باید محصولات را با استفاده از ژل الکتروفورز تفکیک کرده و در نهایت با توجه به سایز قطعه مورد نظر که پرایمر اختصاصی آن طراحی شده، محصول و یا Tm اختصاصی مربوط به ژن هدف را مشخص نمود.
در موارد کاهش غلظت RNA لازم است میزان رسوب RNA را با استفاده از ترکیبات دیگر که روند رسوب آن را بالا می برد افزایش داد. برای انجام این فرآیند در مرحله رسوب RNA با استفاده از الکل، بهتر است از الکل ایزوپروپانول غلیظ استفاده شود . در این مرحله علاوه بر ایزوپروپونال، ترکیباتی از کربوهیدرات های شاخه دار می تواند در به دام انداختن RNA و افزایش رسوب کمک کند. علاوه بر این کاهش دما، می تواند منجر به افزایش رسوب RNA شود. به همین منظور بهتر است پروسه انکوباسیون در ۴ درجه سانتی گراد را پس از اضافه کردن ایزوپروپانول طولانی تر نمود.
خدمات مطالعات مولکولی

خدمات مطالعات مولکولی

آدرس: تهران – اتوبان چمران جنوب – خیابان باقرخان غرب – پلاک ۷۶ شرکت دانش بنیان بافت و ژن پاسارگاد
لطفاً قبل از مراجعه حضوری هماهنگی های لازم را انجام دهید.
۰۲۱۶۶۱۲۱۹۷۶ – ۰۲۱۶۶۱۲۱۹۸۱–۰۲۱۶۶۱۲۱۹۸۳ ۰۲۱۶۶۱۲۱۹۸۸–۰۲۱۶۶۱۲۱۹۹۲ – ۰۲۱۶۶۱۲۱۹۹۷

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}