
ارائه خدمات مولکولی با پیشرفته ترین دستگاه ها و مجرب ترین اساتید
برخی از تکنیک های ارائه شده در هیستوژنوتک
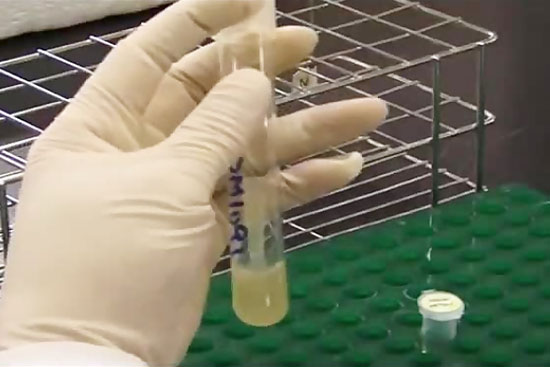
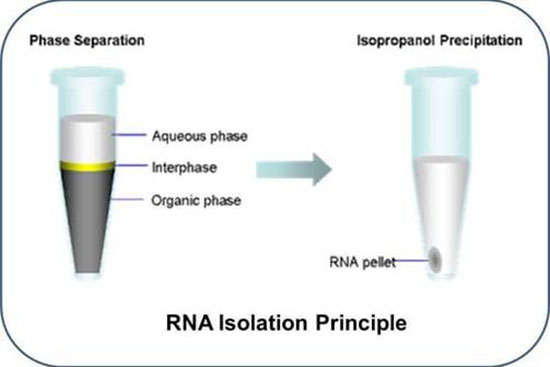
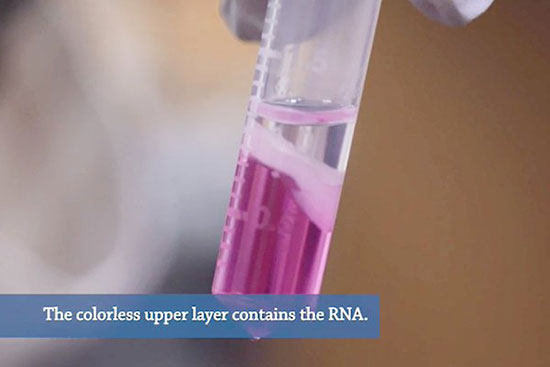
- استخراج RNA از بافت و سلول به دو روش کیت و دستی
- سنتز cDna
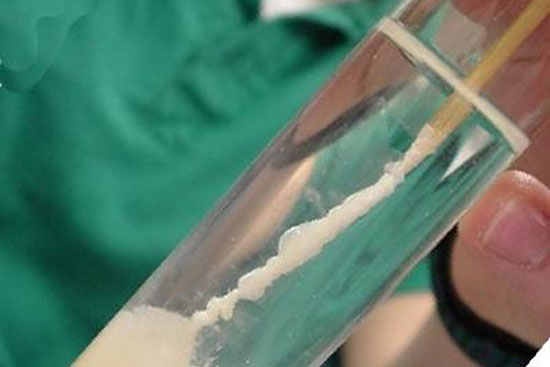
- استخراج DNA ژنومیک از خون، بزاق به صورت دستی و با استفاده از کیت
- طراحی پرایمر جهت بررسی بیان ژن
- طراحی پرایمر جهت بررسی میکرو RNA
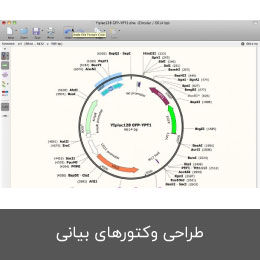
- طراحی وکتورهای بیانی
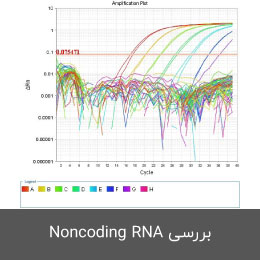
- طراحی پرایمر جهت بررسی Noncoding RNA (میکرو RNA، LncRNA)
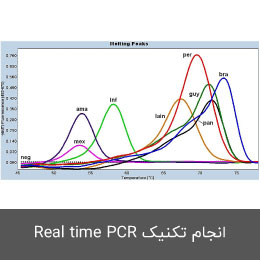
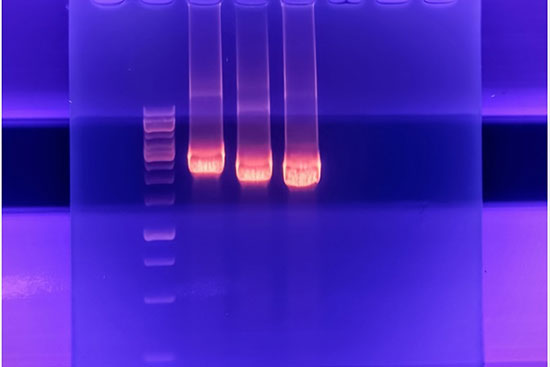
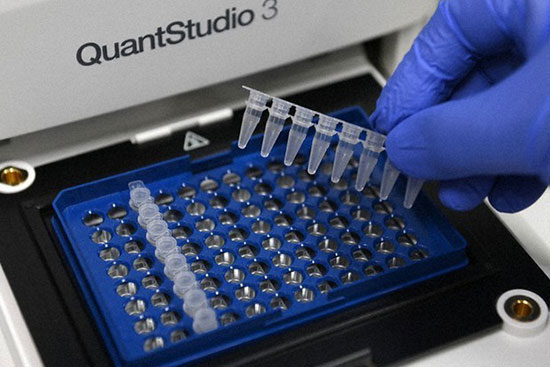
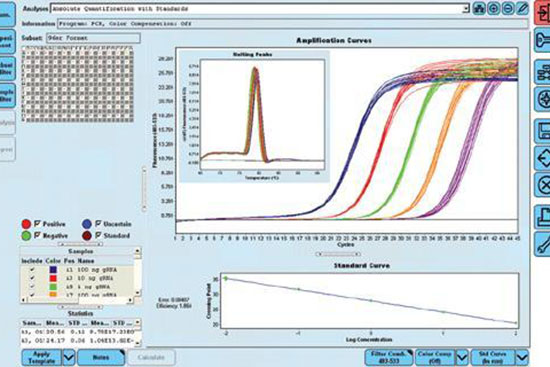
- انجام تکنیک Real time PCR (ریل تایم پی سی آر)
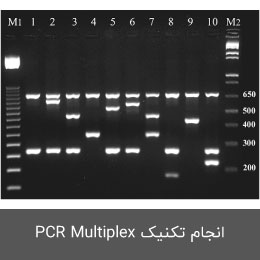
- انجام تکنیک های Multiplex PCR ، ARMS-PCR و Nested PCR </a
- بررسی متیلاسیون در ژنوم به روش BSP و MSP
- بررسی پلیمورفیسم و جهش ژنی به روش RFLP، Nested PCR،ARMS وتوالی یابی ژنوم
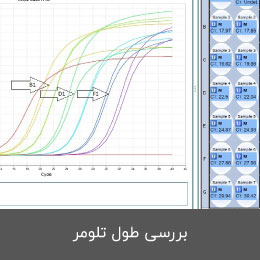
- بررسی طول تلومر با استفاده از روش PCR کمّی
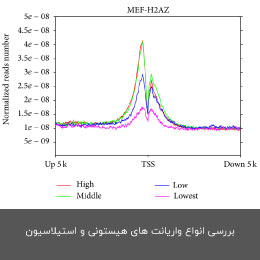
- بررسی انواع واریانت های هیستونی و استیلاسیون
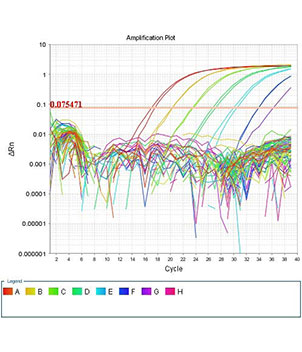
پیشرفت های دو دهه اخیر در زمینه بیولوژی مولکولی موجب ارتقاء کارآیی و تکامل هر چه بیشتر آزمایش های وابسته به DNA در علوم پزشکی گردیده است.
بحث عمده در زیستشناسی مولکولی، استنباط برهم کنش بین سیستمهای درون سلولی ( از جمله برهمکنشهای RNA ،DNA و پروتئین سازی) و چگونگی تنظیم این برهمکنشها می باشد که مورد بررسی قرار میگیرد.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}